INTRODUCTION
There are some things that never change. One of them concerns Pyloric Stenosis of Infancy (PS). It has no known cause! It was 60 years ago when I saw my first case. This despairing mantra has not stopped the regular accounts of the curious clinical features, all repeated to no obvious avail. Causation itself, strangely, in modern times, has excited little interest.
The clinicians who first encountered PS, with only clinical features to guide them, had no such reticence. Freund in 1903 found a hydrochloric acid content in excess of normal and considered it to cause “spasticity” of the pylorus. [1] Berend in 1910 found the volume of vomitus greatly exceeded the volume of feed-intake, indicating that gastric secretions were greater than normal. [1]Others agreed that too much acid was involved and alkaline powders were given with apparent good effect. [1], [2]
In 1911, in an M.D. thesis on Infantile Hypertrophy of the Pylorus (Glasgow University), Dr. D.L. Carmichael confirmed hyperacidity in PS. He also showed that Acid entering the duodenum made the sphincter contract. Nutritious enemata helped produce a long-lasting cure (the less the baby is fed-the better) and advocated the use of alkaline gastric infusions in treatment. Acid solutions left the stomach most slowly. Fixed outlet obstruction at the postmortem was due to the formalin pickling effect. At operation, it was always possible to pass a No. 10 catheter across the lumen. Obstruction in life was due to hypertrophy plus sphincter contraction. It was not a debate between “spasm” and muscular thickening-it was both! Sphincter hypertrophy was the cause since the histological arrangement was maintained. It was not a tumour! [3]
The earliest clinicians also recognized that an enhanced appetite was present before the vomiting started and, strangely, a reduction in feed was helpful in obtaining a long-lasting cure. [1], [2] Feeding progressed the condition.
In recent years, providing titratable acidity is used, hyperacidity has been confirmed [4] both before and after successful pyloromyotomy. [5] When acid enters the first part of the duodenum it causes repeating sphincter contraction and sphincter work hypertrophy is the natural outcome. [6]
Gastric outlet obstruction itself further increases acid secretion [7] and so it goes on. Prof. Dodge’s demonstration that pentagastrin stimulated hyperacidity in puppy dogs created the condition, which seemed to settle the matter. [8] One large problem however still remained. How could we explain, as we must, the extraordinary time-sensitive features?
How the hyperacidity theory and/or overfeeding explains 6 cardinal features
1. When tested under ethical conditions (i.e., with a nasogastric tube for other reasons) premature baby boys secrete much more acid than matched females. [9]
2. Gastric acidity in normal development peaks at around 3 weeks-the times when most PS babies present.[10]
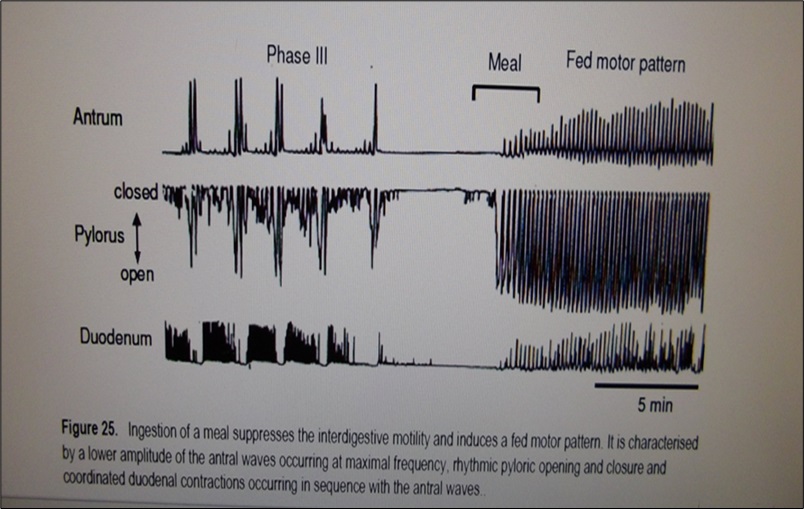
3. Pyloric sphincter contraction is most frequent and of greatest amplitude after feeds (Fig. 1). [11]
4. Temporary reduction of acidity- either naturally with time or by the temporary prescription of acid-blocking drugs is associated with an enduring cure in milder cases. [12] All pediatricians who look after PS babies will know that the condition does exist in a mild form which can self-cure with or without treatment. The passage of time may be all that is required.
5. When babies who develop PS have been fed 3 hourly instead of 4 hourly, they present at an earlier age. [13]It is credible to suppose that first-born babies are fed more frequently by their anxious novice mothers, especially when vomiting begins and their babies are even more hungry and dehydrated than before. The first-born effect is only apparent from the 3rd. week of life onwards when anxious overfeeding has had time to work. There is no first-born phenomenon in the first 2 weeks. [13] While the novice mother may not relatively overfeed a normal child- i.e. to initiate the condition- it is difficult to imagine she will not overfeed her vomiting baby in her anxiety and thus add to the progression of the condition. A definitive study has not been done and would be difficult to objectify.
6. The potential to secrete acid is transmitted as a multifactorial genetically determined mechanism and gives rise to familial occurrence. The focus on genetic control for acidity has yet to be determined. It is of interest that both PS babies and the adult equivalent of a duodenal ulcer are associated with a preponderance of blood group O and classically retain a good appetite. [14], [15]
Why hyperacidity peaks at 3 weeks of age and why PS may self-cure as time passes
In 1974 we reported the phenomenon of neonatal hypergastrinemia.[16] The fasting gastrin, though higher than the maternal gastrin on Day 1, rose rapidly to become 2-3 times higher by Day 4.
The newborn stomach is usually neutral or alkaline due to swallowed amniotic fluid. By Day 4 the neonatal stomach is known to have become acidic. Thus, gastrins and acidity rise together during this time. We speculated that the adult negative feedback between gastrin and acidity had not yet developed at that time. In a sense, the normal newborn baby was acting like a mini-Zollinger-Ellison syndrome.
Such a system predicts a temporary peak level in acidity and gastrin when negative feedback matures. It also predicts a fall in both levels thereafter when both come under mutual restraint.
In a classic paper in 1969, Dr. Mona Agunod showed that acidity did rise to a temporary peak at around 3 weeks of age in normal development before falling again. [10] Others have confirmed that there is a similar parallel temporary peak in gastrin levels around that time. [18], [19]
In this way, the presentation at 3 weeks due to peak acidity, self-cure with time due to falling acidity, luminal enlargement, and greater baby control of feed intake were explained.
Others more recently have added to the evidence that an early impaired negative feedback exists not only in man but in beagle puppy dogs.
1. Pentagastrin has no acid-secreting effect on Day 1 of the birth of the human baby. The gastric mucosa is already being maximally stimulated. [17]
2. High fasting gastrins exist in the early days with no post-feed-gastrin increase but in later weeks when fasting gastrins fall, there is a post-feed-gastrin increase. [18], [19] Only when gastrin is under feedback control, can food produce a gastrin increase.
3. Beagle puppies studied from birth to 5 weeks of age showed that they also had neonatal hypergastrinemia. Temporary peak gastrin occurred on Day 7 and then fell to Day 18. Similarly, peak acidity occurred on Day 7-and pentagastrin increased acidity only after the 9th. postnatal Day when feedback had matured.[20]
4. Most interestingly in puppies, antral contractions rose from 1 every 5 minutes to 11 every 5 minutes during the period when acidity was increasing. [20] Such a phenomenon no doubt will also be associated with more frequent acid-stimulated sphincter contraction.
There are many physiological consequences of immature negative feedback:
1. Gastric acidity is artificially increased when the neonatal gut is vulnerable to bacterial attack without necessarily leading to hyperacidity problems in later life.
2. The unrestrained gastrin increase in the first few weeks of life is known to cause hyperplasia of the gastric mucosa. Thus acid-secreting capacity is increased and the neonatal gut is prepared for an independent digestive life.[21]
In one sense PS is the price a small minority ( about -0.5%) pay for the safe bacteria-free passage to the independence of the great majority. An enhanced parietal cell mass, male sex, and possibly an overanxious first-time mother will add to this condition.